The FDA recently approved two new treatments for an ancient scourge, sickle-cell anemia. Vertex Pharmaceuticals’ Casgevy and Bluebird Bio’s Lyfgenia are not prescription drugs, but processes that genetically modify a patient’s own cells before they are then returned to the patient.
The treatments could be groundbreaking, but with them come multiple questions.
What is sickle-cell anemia, and how has it been treated until now?
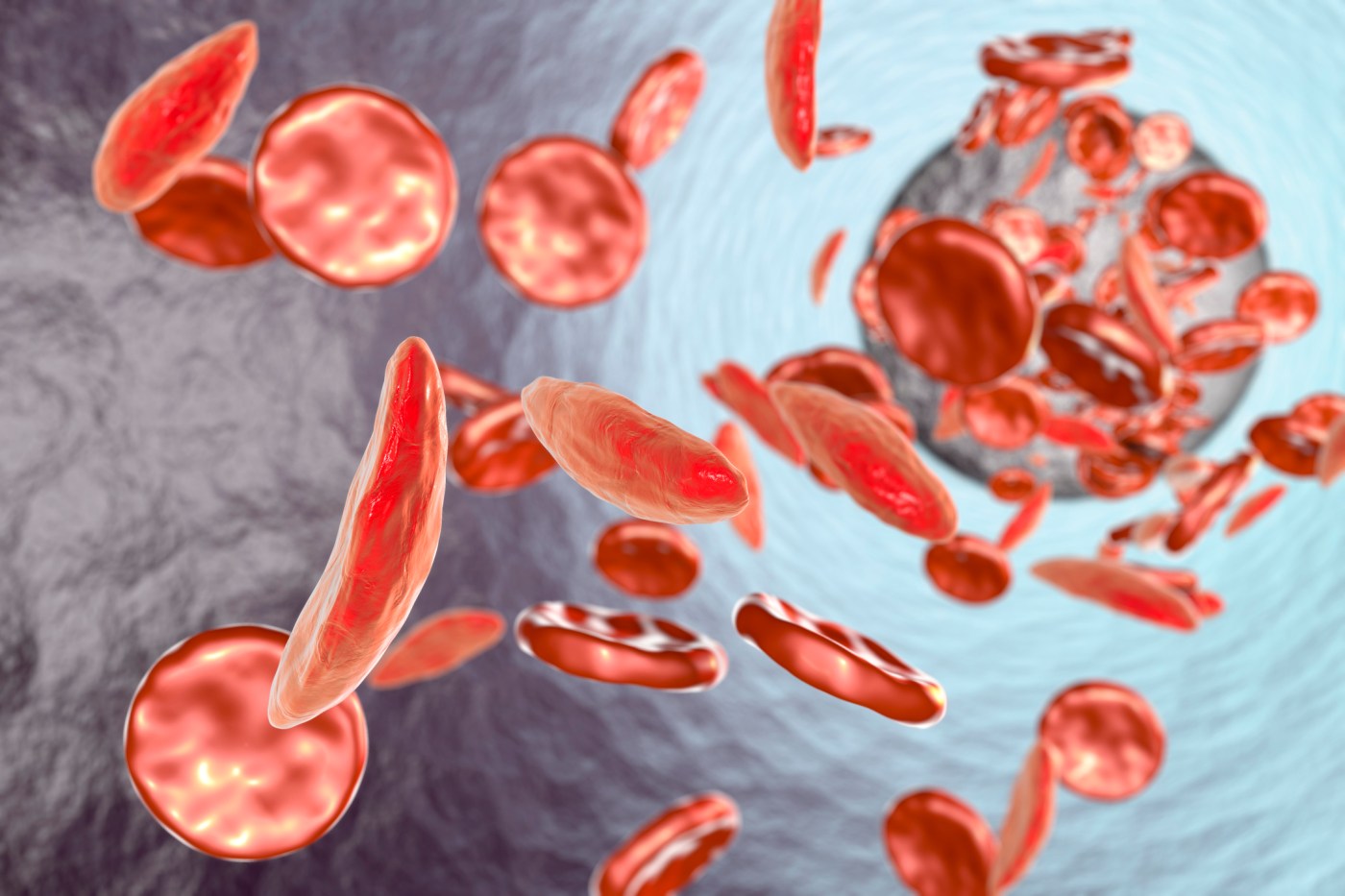
Sickle-cell is a genetic disease whose sufferers’ hemoglobin (the blood component that carries oxygen) causes some of their red blood cells to form a sickle-like shape. The misshapen cells have a much shorter lifespan than normal red blood cells, giving rise to anemia as the body struggles to replace them. They also jam in microscopic blood vessels, causing excruciating pain and damage. Although most sickle-cell patients in the U.S. now live to adulthood, they still have significantly shorter lives than the general public. In poorer countries, sickle-cell anemia is often fatal in childhood.
The American Society of Hematology’s first-line drug for sickle-cell works by increasing production of fetal hemoglobin (HbF)—a different form of hemoglobin produced by unborn babies and infants (with or without sickle-cell) in their first few months. HbF doesn’t sickle. Some patients benefit greatly from it. Yet the drug, hydroxyurea (also called hydroxycarbamide), has not proven to be as potent as its advocates once hoped, and it carries its own risks: The American Society of Health-System Pharmacists describes it as a “highly toxic drug,” a “presumed human carcinogen” known to be hazardous in pregnancy and associated with “possible severe, sometimes life-threatening or fatal, adverse effects.” Even so, many sickle-cell patients take hydroxyurea faithfully: For them, it is still better than what they endure without it.
Two newer drugs, Pfizer’s Oxbryta and Novartis’ Adakveo, have addressed sickle-cell more directly. Both carry extreme price tags, with a month’s supply of Oxbryta listing for $14,270, and each is meant to be taken for life.
Blood transfusions address anemia directly, and they help during the painful “vaso-occlusive” crises sickle-cell patients dread, but:
- Their supply is limited.
- Each transfusion carries a small, but non-zero, risk of transmitting blood-borne diseases like HIV and hepatitis C.
- Each time a patient receives a transfusion, he or she risks developing antibodies that will make future searches for compatible blood more and more difficult.
The definitive treatment to date has been a bone marrow transplant. Because a successful bone-marrow transplant effectively replaces the body’s entire blood production system with that of a healthy person, it also cures sickle-cell anemia. (The first patient to be cured of sickle-cell anemia with a bone-marrow transplant was technically being treated for leukemia, but the transplant cured both the leukemia and the sickle-cell.)
Yet a bone-marrow transplant is a long and harrowing journey, with a mortality rate approaching 10 percent. Even attempting the transplant requires agonizing rounds of chemotherapy to destroy the existing hematopoietic (blood production) system.
How do the new treatments compare to the old ones?
The FDA describes Casgevy and Lyfgenia as gene therapies. They are, but the specifics matter: These gene therapies modify cells taken from patients, so that those cells can then be given back to the same patients as an autologous (from the patient’s own cells) bone-marrow transplant. The methods differ between the two treatments. Casgevy manipulates DNA directly with the CRISPR technology that won a 2020 Nobel prize for making gene editing a “cheaper, faster, easier” process. This treatment manipulates DNA directly, switching off the gene responsible for preventing fetal hemoglobin production—in other words, moving the body back to making it. By comparison, Lyfgenia uses a modified virus to inject genes for something like adult hemoglobin into the patient’s cells.
Since these treatments produce a slightly modified version of a patient’s own cells, scientists believe the modified cells and the native cells should be similar enough that they view each other as friends, not strangers. Since GVHD is one of the main problems with bone-marrow transplants, eliminating it would substantially reduce the risk of seeking to cure sickle-cell disease this way.
Does this mean curing sickle-cell is easy now?
Not really. The months-long hospitalization, including time spent in isolation with effectively no immune system, remains. The chemotherapy to pave the way for the autologous transplant also remains. The transplant still occasionally fails—about 10 percent of the time for matched family members. In the small clinical trials for Casgevy and Lyfgenia, none of the transplants initially appeared to have failed. However, Lyfgenia’s FDA approval does call for repeated genetic testing to follow up whether its edits worked as hoped.
While the technology appears promising, researchers have tested it only in small clinical trials. The CRISPR researchers believe their technique will only edit DNA at the correct location, but they could be wrong. And editing DNA at other sites in the genome could have unknown consequences.
What do these treatments cost?
There’s the $2 million question—literally. Estimates range from roughly $2 million to more than $3 million, based on a theoretical average lifetime cost of treating sickle-cell of $4 million: Each patient’s treatment is made individually from his or her own native stem cells.
This raises the usual questions of how treatments are priced, and an even more basic question: What good is an effective treatment if patients can’t get it? Most sickle-cell disease afflicts people in the developing world, only a tiny fraction of whom can hope to receive a bone-marrow transplant—much less one genetically modified to give better chances of success.
Beyond their financial cost, these treatments trade a familiar course of treatment for another, unknown course. The current course of treatment—working to avoid pain crises for the patient, staying on medications that reduce them, and managing them when they occur—gives way to a future without pain crises, but with risks related both to the preparation for the autologous transplant and to the transplant itself. Chemotherapies are inherently toxic drugs and often pose a risk of cancer themselves; since stem-cell transplants work best in younger patients, setting events in motion that may lead to cancer 30 or 40 years in the future may mean much more than it would when giving chemotherapy to a patient well into adulthood. They also often render patients infertile.
Both Casgevy and Lyfgenia received approval on the strength of small studies. Two of the 45 patients in the Lyfgenia trials developed blood cancers—hinting at the wrong-site problem mentioned above—and the FDA states that patients receiving either treatment should have ongoing monitoring for them.
So what does this all mean?
In the short term, it means hope for patients with severe sickle-cell disease (and excellent insurance), especially those without access to the type of donor who would have posed comparatively lower risk for post-transplant complications—like siblings and parents.
Many new treatments offer the most relief to patients with comparatively milder forms of a disease, but the opposite holds here: patients with less severe sickle-cell probably won’t roll the dice on a bone marrow transplant of any type, whereas those with severe disease may.
In the long term, this opens the door to a world of genetic modification, which will require society to consider several hitherto-unexplored questions.
What new ethical questions does this raise?
Just as oncologists place great emphasis on whether a tumor has even once gone somewhere other than where it started, humankind has now even once succeeded in editing its own genetic code. It is difficult to overstate the significance of this development.
CRISPR technology can also be used to edit germline cells, meaning (to oversimplify a bit) that we now have the technology to make heritable changes—changes that will be passed down through generations. Calls have already gone out online to regulate human genetic modification, limiting it to treating intractable diseases like sickle-cell anemia.
But the question will soon arise: Once we start editing the human genetic code, where—and how—do we stop?
Sickle-cell seems to be an easy call: Plainly the human body’s design calls for functioning red blood cells. Other genetic illnesses carry the same argument—would we deny treatment to a person with Tay-Sachs, muscular dystrophy, Huntington’s?
But what’s the best way to prevent these same technologies from being misused? Will society refuse to let them be deployed to make our children more intelligent, longer-lived, tall-blonde-and-blue-eyed—just as long as we’re willing and able to pay?
If we say, “We’ll regulate it,” still more questions arise: Who does the regulating? And how? When the inevitable rogue geneticist modifies germline DNA, how exactly do we propose to deal with the babies that come from it?




Please note that we at The Dispatch hold ourselves, our work, and our commenters to a higher standard than other places on the internet. We welcome comments that foster genuine debate or discussion—including comments critical of us or our work—but responses that include ad hominem attacks on fellow Dispatch members or are intended to stoke fear and anger may be moderated.
With your membership, you only have the ability to comment on The Morning Dispatch articles. Consider upgrading to join the conversation everywhere.